You know you should be using the FDA 2011 Process Validation (PV) Guidance, so why aren’t you?
Bad habits interrupt our lives and prevent us from accomplishing our goals. Some, like smoking, jeopardize our physical health. Others, like self-criticism, take a heavy mental toll. All bad habits waste time and energy. So, why do we cling to them?
Change is hard, uncomfortable even. Habits are so ingrained in our brain, we perform them automatically, even when we stop benefiting from them.
The same holds true for detrimental work habits. You know you shouldn’t check your email every five minutes or gossip at the water cooler, but you do. While some bad habits can zap your productivity, in high-risk environments, such as pharmaceutical manufacturing, they can be deadly.
Yet time and again, I see process people (R&D, production, engineering, QC, QA) clinging to the FDA’s 1987 process validation (PV) guidance (i.e., IQ/OQ/PQ) as a framework for technology transfer rather than adopting the risk-based 2011 version. I frequently speak at seminars and conferences nationwide about the subject of process validation. When I query the crowd, virtually no one raises their hand to confirm they are using the 2011 guidance. This is shocking to me. After all, it’s been five years!
Whether they avoid using it out of habit or hubris (“we know our processes better than regulators”), it is, in my opinion, like playing with a loaded gun. Before I explain why, let’s define technology transfer, and why it is such a critical step in the lifecycle of a drug (figure 1), or, more importantly, in the processes that produce drugs.
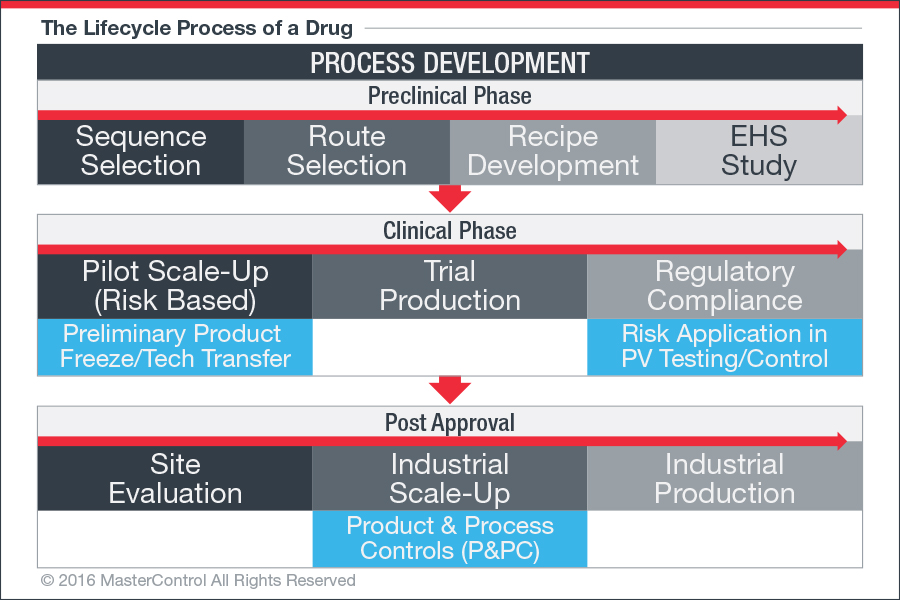
Figure 1: For a typical research-based pharmaceutical company, drug discovery and development can be broken down into distinct stages.
Technology Transfer: 10,000-Foot View
Technology transfer is now specifically an element of design and development for pharmaceutical drugs; it’s governed by ICH Q10. In the pharmaceutical industry, technology transfer can mean many things:
- The processes that are needed to transfer all of the information and technology necessary to manufacture a drug product consistently
- The process of taking a drug from R&D to full-scale commercialized product
- The process by which a developer of a drug makes the drug available to a commercial partner (e.g., CMO, compounding pharmacy, or supplier) that will exploit or manufacture the technology
- The process of moving a drug from one process train to the next—e.g., from base manufacturing (API or other ingredients) to cumulative manufacturing (mixing/blending); from cumulative manufacturing to product configuration (tablet, capsule/liquid); and from product configuration to packaging and labeling
- The process by which a developer of a drug transfers the development and manufacturing data of a drug from an existing manufacturing site to a new manufacturing site or from one facility to another
- The process of incorporating post-marketing feedback which instigates changes in the drug development and manufacturing processes
- The influence of new technologies such as cell therapeutics and biologics license application (BLA) products
Technology transfer and validation are much larger concepts than can be covered here. For the purpose of this article, I will focus my discussion on the application of risk, as it relates to technology transfer, using the FDA’s PV 2011 framework.
The “New-ish” PV Model: What’s Changed?
The “new” guidance advocates a three-stage, phase-gate approach to validation. It aligns process validation activities with the product lifecycle concept, as well as with existing guidelines ICH Q8, ICH Q9, and ICH Q10. Product lifecycle in the life science industry refers to the entire process of product development, from design to commercialization.1 Although explained in the guidance as “discrete stages,” some activities like risk assessments can occur in multiple stages or overlap between stages. The guidance describes the three stages as follows (figure 2):
- Stage 1—Process Design: The commercial-manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities (i.e., planning).
- Stage 2—Process Qualification: During this stage, the process design is evaluated to determine whether the process is capable of reproducible commercial manufacturing (i.e., application of control measures; identifying attributes in your process that would trigger change or even stop the process).
- Stage 3—Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control (i.e., risk-based process and planning control or P&PC/ statistical quality control or SQC).2
This new “PV-as-a-lifecycle” concept—as opposed to PV being a three-batch static event—requires a huge shift in thinking for industry, which probably accounts for the poor adoption, but it’s just one of many changes.
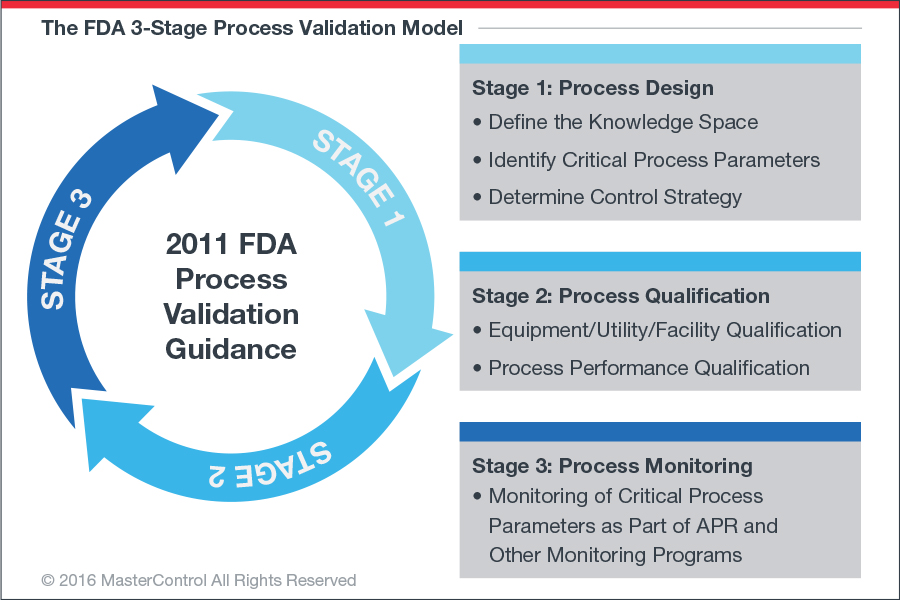
Figure 2: The new “PV-as-a-lifecycle” concept requires a huge shift in thinking for industry.
Scientific Evidence Required
The 1987 guidance emphasized the assurance of quality through sampling and testing, and then documenting that your process does what it purports to do—pretty straightforward. Under the 2011 PV guidance, quality is demonstrated, not by sampling and testing, but by design (QbD) and control. Although many see this as unnecessarily complicating an already complicated process, I disagree. The IQ/OQ/PQ method requires an enormous amount testing, resulting in data overload (the golden rule of threes).
Using the updated guidance, you can legitimize minimal testing with minimal risk and maximum control. This is because, in all reality, the second stage in PV 2011 incorporates IQ/OQ/PQ. So, hypothetically speaking, if you do a good job during stage one, you could realistically skip stage two. And by good job I mean you can prove that your design is robust, controls risk, and is statistically sound. The caveat is—and it’s a big one—you must be able to demonstrate that you have a scientific understanding of your process (or processes) and how it effects your product (i.e., risk). Admittedly, this is no small feat. For years we’ve been told that process validation is a documentation exercise, and now the FDA wants us to approach it scientifically. A 30-year-old habit is hard to break.
“Stuff Happens”
While these changes and concepts may seem revolutionary, they aren’t really. W. Edwards Deming suggested implementing this level of process validation control nearly three decades ago. Unfortunately, the industrial public didn’t listen. Instead, they sent him packing to Japan, and the United States deployed compliance as overkill using IQ/OQ/PQ. Many still aren’t listening.
When I ask advocates of the 1987 version why—if the original PV guidance works—we continue to see processes fail during manufacture, the usual response is: “stuff happens.” It sure does. Industry publications are filled with examples of drug failures due to poor technology transfer and validation processes. A large India-based pharmaceutical company has had several recalls this year, and three of its manufacturing plants have been targeted in an FDA warning letter. That’s just one example. “Stuff happens” is really just code for “process variability,” and we have to get better at predicting and controlling it. FDA PV 2011 can help.
![]()
As the Saying Goes…
There is an old saying that things on the bench don’t always work on the floor. This is true now more than ever thanks to rapidly expanding technologies like laboratory developed tests (LDTs). Very basic technology transfers—e.g., batch vs. continuous blending of complex formulations—are no longer the norm. As product/process complexity increases, so must the risk assessment and validation efforts of the drug maker or CMO. A 30-year-old PV framework, which doesn’t even address risk, simply won’t cut it.
Risk has become the cornerstone of standards and regulations worldwide. All of the new ISO standards require adherence to risk; it is explicit. While GMP regulations do not explicitly require the implementation of risk management principles, it is certainly implied. If an auditor (FDA or customer) finds out you are using the old PV orientation, you’re toast (i.e., out of conformance). No excuses.
What Are You Waiting For?
I would like to challenge each and every one of you to start implementing PV 2011 strategies into your validation and transfer technology processes—today! Don’t put it off until tomorrow, next week, next month, or until things “slow down.” There is no better time than right now.
Start with product realization planning. (Per ISO, “product realization is the term used to describe the work an organization goes through to develop, manufacture and deliver the finished goods or services.”) This will allow you to focus on control planning, deploying objectives, and assessing risk. Your suppliers should play a major role in providing testing data that qualifies the equipment, utensils, and facility; this is factory acceptance testing (FAT). You, as the manufacturer, should evaluate the normality and level of control in process design and extend any necessary site acceptance testing (SAT). Note: FAT data should drive how aggressive the SAT data is going to be in the scale-up.
This should all be done to provide statistical quality control (SQC). The SQC data, through product performance qualification (PPQ), should translate to statistical process control (SPC). If you are doing the right things to maintain controls, whether human or technological, good risk-based decision making will ensue.
References:
- “The Importance of Product Lifecycle Management Solutions in the Life Science Industry,” MasterControl, accessed September 12, 2016, http://www.mastercontrol.com/plm/plm_solution.html
- “Guidance for Industry: Process Validation: General Principles and Practices,” FDA, Jan. 2011. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070336.pdf
This feature story can also be found in the November/December 2016 issue of Pharmaceutical Processing.
Follow us on Twitter and Facebook for updates on the latest pharmaceutical and biopharmaceutical manufacturing news!



