In recent years there has been a significant increase in the number of warning letters referring to ‘data integrity’ and ‘sterility assurance’ in relation to environmental monitoring.
Abstract
In recent years, the FDA response to companies with insufficient proof of control over their production has become more aggressive, demonstrated by the rise in 483s and warning letters issued. Topics related to environmental monitoring programs are included in the top 15 reasons 483s are issued. By becoming aware of the regulations and reasoning behind 483 distribution, other pharmaceutical companies can become better prepared against making the same mistakes.
Introduction
The FDA Committee has increased warning letter distribution dramatically in recent years. Notably, there has been a significant increase in the number of warning letters referring to “data integrity” and “sterility assurance” in relation to environmental monitoring (EM). The increase is attributed to a dramatic change in the FDA regulator approach to infraction handling. Those of significance to the 2016 drug sector and relating to EM are highlighted in blue in Table 1.
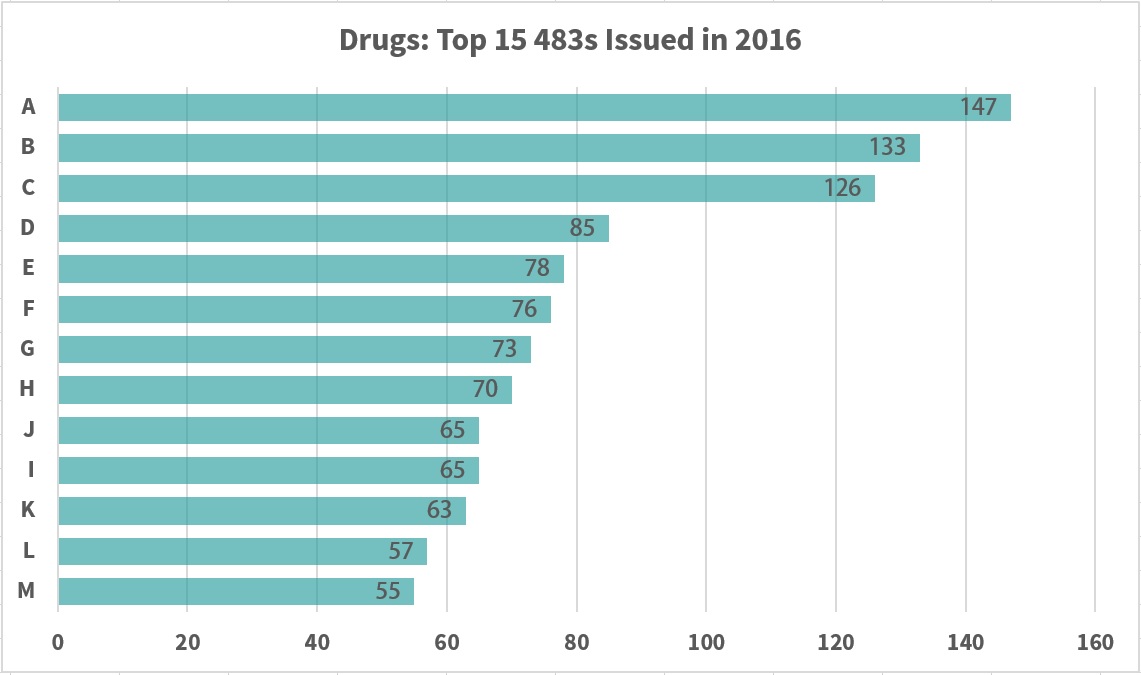
Information from https://www.fda.gov/ICECI/EnforcementActions/ucm531890.htm#Drugs Inspections ending between 10/1/2015 and 9/30/2016

The FDA fully expects that product bioburden be assessed and evaluated. CFR 211.113(b) Control of Microbiological Contamination states that the “appropriate written procedures designed to prevent microbiological contamination of drug products purporting to be sterile, shall be established and followed.” Such procedures should include validation of all aseptic and sterilization processes.
Warning Letters
There are several common violations found by FDA compliance officers, including a lack of quality control system, failure to establish and follow written procedures (for quality control and process validation), failure to utilize cleaning practices in order to prevent contamination or malfunction, failure to exercise control over computerized access and data, and failure to investigate violations found.
Written Procedures
In the Mylan Laboratories Limited 8/6/15 and Sato Pharmaceutical Co., Ltd. 2/2/17 warning letters, it is stated that each company did not establish or follow written procedures. These procedures should be “designed to prevent microbiological contamination of drug products purporting to be sterile,” as per 21 CFR 211.113(b).
In response, the inspector requested a risk assessment “describing process failure modes, full sterility history (e.g., sterility testing, media fills), and all actions taken to evaluate and address the acceptability” of the product.
Investigation
In the Formatech Inc. 2/10/11 warning letter, the failure to provide a thorough batch failure investigation, including its components, is addressed. Product specifications must be met despite whether the batch has already been distributed (21 CFR 211.192). The firm “routinely failed to thoroughly investigate and identify root causes when environmental monitoring data exceeds the action limit.”
As a response, the firm hired a consultant to address the infraction by assessing environmental data and repairing the facility. However, the response was insufficient due to inadequate investigation of “disinfectant procedures, frequencies, and preparation.”
Several other infractions were noted including:
- The disinfection program was a failure due to “insufficient use of sporicidal agents”
- Environmental data from 2008 to 2010 was evaluated and showed a “lack of comprehensive investigations when mold and bacteria were identified,” which went above action levels
- Due to the process relying on manual manipulations (involving personnel in close proximity to the product), the poor demonstration of environmental control could pose a significant risk to contamination
It is essential that the environmental monitoring program is continually maintained throughout the aseptic processing facility. Documentation of proper EM practices is essential to ensure successful investigations.
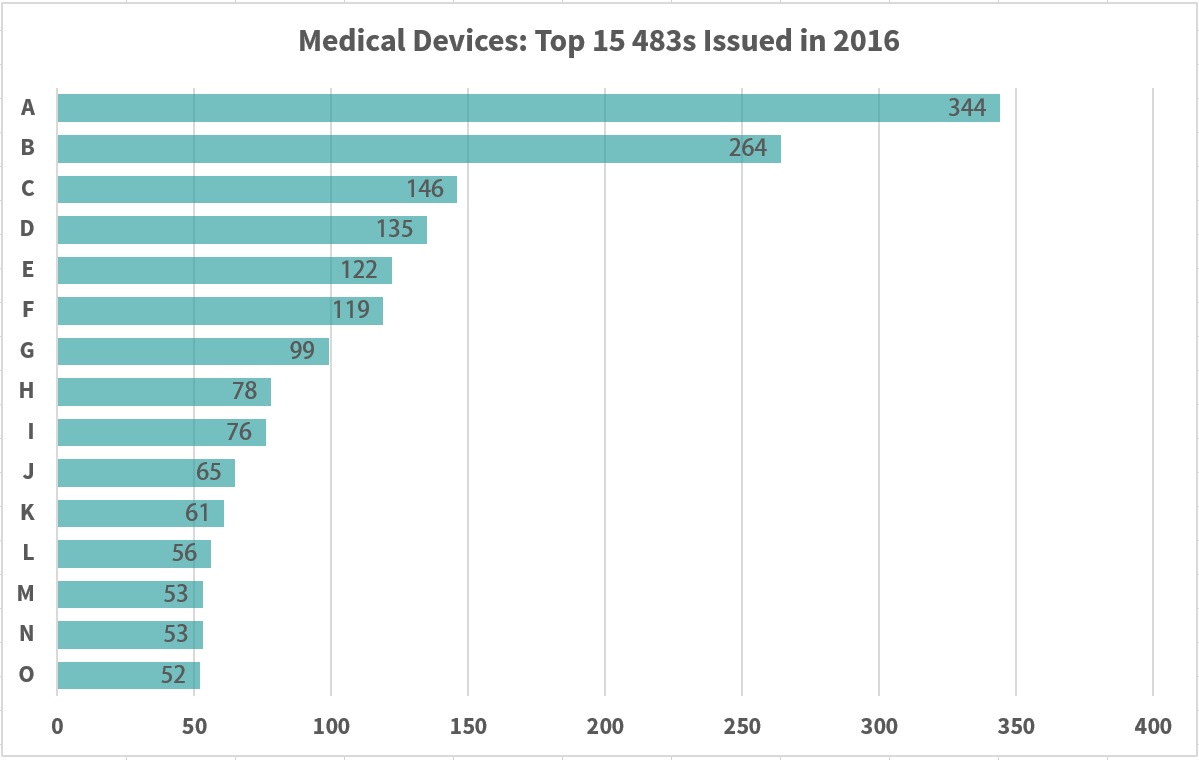
Information from https://www.fda.gov/ICECI/EnforcementActions/ucm531890.htm#Devices Inspections ending between 10/1/2015 and 9/30/2016
Control Systems
The warning letter to Sanofi Aventis Deutschland GmbH dated 2/09/11 stated there was a failure to establish separate or defined areas necessary “to prevent contamination or mix-ups during aseptic processing” (21 CFR 211.42(c). The following was noted:
- The environmental monitoring program did not assure “environmental contaminants are reliably detected”
- Sample collection from personnel “from left and right hands on alternate days” was “unacceptable”
- The SOP failed to include instructions for the “location and duration of samples collected in the critical aseptic processing area”
- An adequate environmental monitoring program should be established, and “capture meaningful data,” in addition to acting as “an early warning system to detect possible environmental contaminants that may impact the sterility of drug products”
In the Sato Pharmaceutical Co., Ltd. 2/2/17 warning letter, the inspector noted that the company failed to “establish an adequate system for monitoring environmental conditions in aseptic processing areas” (21 CFR 211.42(c)(10)(iv).
Routine personnel monitoring and surface sampling, and air quality data were not shown to occur in any capacity, and it was requested for the company to provide a reassessment and CAPA of the environmental monitoring program to “ensure it supports robust environmental control” with guidelines such as:
- Justifying sampling locations, and associated action and alert limits
- Ensuring all locations are sampled at appropriate frequencies, with special emphasis on implementing routine sampling of aseptic processing room surfaces
- Defining circumstances under which investigation of an adverse trend or out-of-limit result is triggered, as well as appropriate responses to such occurrences in order to promptly address contamination hazards
Not only is it important to have an EM device that provides accurate and reliable data, but it is also imperative that the program dictating when and where monitoring takes place is proven to ensure product quality.
Incomplete Data
Failure to include complete data derived from all tests necessary to assure compliance was noted in Cadila Heathcare Limited’s letter on 6/21/11. The importance of ensuring compliance with established specifications and standards is noted in 21 CFR 211.194.
In particular, the infraction included microbiologists reporting plates as having no CFUs when two plates actually contained one each. Due to the action limit of the plates’ sample locations, an investigation was required as per the company’s SOP.
In addition, investigators frequently specify the need for audit trails and access controls for computerized data collection (see 21 CFR Part 11). This is noted in several recent warning letters, including BBT Biotech Gmbh 5/16/16.
Microbiological Contamination Control
It must be established through detection testing that the product meets microbiological quality standards (see USP 37, <62> Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms).
A “specified” microorganism has several elements that require evaluation on a case-by-case basis for each drug manufacturer. The term refers to microbial contaminants that, based on microbial species, number of microorganisms dosage form, intended use, patient population, and route of administration would adversely affect product safety. Most “specified” microorganisms would pose a threat of patient infection or mortality.
It is no longer possible to release products or monitor the processes (especially aseptically filled sterile products) using microbiological methodologies/techniques developed in the 20th century. There are four types of modern monitoring systems that offer various limitations on personnel interaction with the product.
- In traditional cleanroom production, the presence of people in a Grade A area is allowed, with a mandatory installation of a surrounding Grade B environment
- In open Restricted Access Barrier Systems (oRABS), there is a physical separation of people from Grade A areas, but Grade A air is exhausted into Grade B. oRABs must be installed with a Grade B surrounding environment
- In closed Restricted Access Barrier Systems (cRABS), there is a physical separation between people and Grade A production areas and Grade A air recirculation. cRABS must be installed with a Grade B surrounding environment
- In an isolator system, the production inside the isolator is completely separated from people and air circulation in a Grade A area. The isolator can be installed in a Grade C environment
Of the four options above, only the isolator system is able to offer complete sterility assurance. With increasing human intervention of the system, more risk is introduced and the ability to ensure a sterile final product is lessened.
At most, 10 percent of pharmaceutical companies are able to utilize isolators as part of their production process, with a larger minority using traditional cleanroom techniques and the majority using a form of RABS. Intervention can be mitigated with a microbiological monitoring method that offers advanced sensitivity with real-time results.
The use of outdated microbiological analytical methods (e.g. settle plates) only allows for the detection of one-third the microorganisms present in the production process. As a result, it is not possible to use these methods to completely identify areas of contamination.
With a more reliable method with advanced sensitivity, improved detection of microorganisms in critical environments is possible, and can positively impact product quality.
Environmental monitoring systems are an important part of aseptic processing of sterile pharmaceuticals, in that they aid in the capability to control the presence, distribution and a result, the survival of microorganisms.
Critical areas, such as process waters (deionized, RO and WFI), air and compressed gases, in addition to working surfaces (personnel, gloves, equipment) should be the primary focus in a monitoring program. Early evaluation of the surface, personnel, and additional critical points of the aseptic manufacturing area aids in minimizing corrective action time.
Additional benefits to a strong EM program include undelayed product release, enhanced efficiency and productivity (labor and time), overall cost reduction, and data integrity.
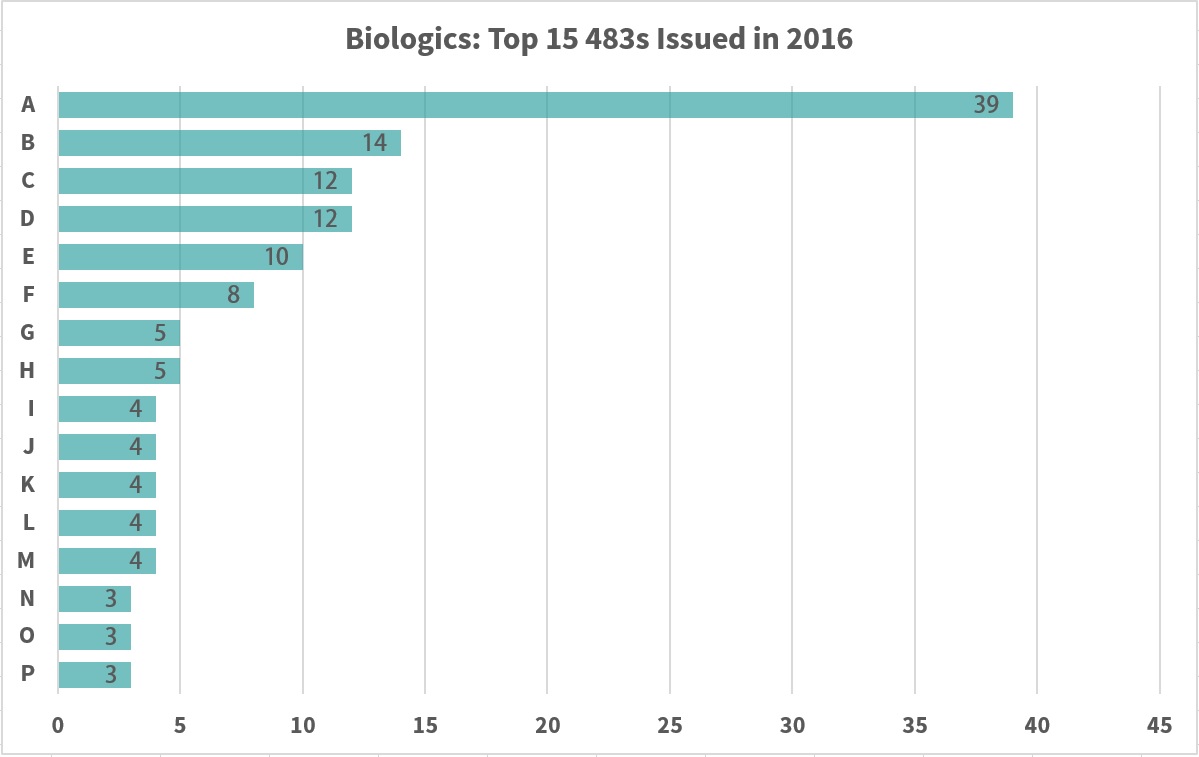
Information from https://www.fda.gov/ICECI/EnforcementActions/ucm531890.htm#Biologics Inspections ending between 10/1/2015 and 9/30/2016
Industry Innovation
The FDA has recognized the need for innovation in the pharmaceutical industry. In 2002, four key initiatives were put forward to instigate this change, including the Critical Path Initiative, Pharmaceutical Quality for the 21st Century – A Risk Based Approach, Quality by Design (QbD) and Process Analytical Technology (PAT).
The collective goal of these new measures is to modernize and improve the quality of pharmaceutical manufacturing processes, and encourage the industry to implement risk-based, continuous, real-time quality assurance.
The desired attributes for a measuring system include:
- Sensitivity at the single-cell level
- Discrimination between viable and non-viable microorganisms
- Ability to detect VBNCs
- Rapid action: as fast as possible (days vs. hours vs. minutes)
- Qualitative and quantitative capabilities
- Ability to identify in case of contamination
- Cheap
- Easy to use and validate
- Robust
- Usable in the manufacturing environment
- Does not contribute to contamination
Full understanding of the production and process needs are paramount to choosing the appropriate monitoring method, which is best handled with a risk assessment. Be aware that different technologies will require specialized implementation.
Different sampling tools and measuring systems will offer flexibility in the application of the appropriate technology for each sampling point. Specific methods should be registered for monitoring critical quality parameters and critical process parameters.
Conclusion
The issuing of 483s has increased significantly, becoming a growing trend of change in the pharmaceutical industry. Many of the cited infractions impact quality control, and by extension, environmental monitoring.
Even more 483s are likely to be dispersed in coming years, and it is best prepared for with a strong understanding of industry standards and a defensible monitoring strategy that offers a complete picture of the production process.
When developing an environmental monitoring program, look to new technologies that offer advanced sensitivity and reliability, with a focus on minimizing human intervention. Choosing to do so will improve sterility assurance and mitigate risk to the final product.
About the Author
Gilberto Dalmaso, Ph.D. has more than 25 years’ experience in pharmaceutical microbiology and sterility assurance. His current work is focused on pharmaceutical microbiology and aseptic processes, microbiological contamination control, and rapid microbiological methods, QbD, and PAT.
Follow us on Twitter and Facebook for updates on the latest pharmaceutical and biopharmaceutical manufacturing news!
